
...
Options for certain conditions caused by cystic fibrosis include:
- Nasal and sinus surgery. ...
- Oxygen therapy. ...
- Noninvasive ventilation. ...
- Feeding tube. ...
- Bowel surgery. ...
- Lung transplant.
Medication
10 rows · Nov 17, 2021 · Cystic fibrosis treatment options The foundations of cystic fibrosis treatment are: ...
Procedures
People with cystic fibrosis may need to take different medicines to treat and prevent lung problems. These may be swallowed, inhaled or injected. Medicines for lung problems include: antibiotics to prevent and treat chest infections a combination of 3 medicines (Kaftrio) to treat the root cause of cystic fibrosis in people age 12 and over
Therapy
Treatment is broken up into medications, respiratory therapies, nutritional support, and exercise. Medications for cystic fibrosis include constant antibiotics, which can prevent and reduce the frequency of lung infections. Airway clearance or respiratory therapies are aimed at loosening up the thick mucus from the lungs.
Nutrition
Dec 23, 2020 · The inhaled antibiotics used for CF are aztreonam lysine, tobramycin inhalation powder/solution, inhaled colistin, liposomal amikacin, liposomal ciprofloxacin, and inhaled levofloxacin. 39, 40, 41, 42 Inhaled tobramycin and inhaled aztreonam are the two inhaled antibiotics with FDA approval. Liposomal amikacin was approved by the FDA in 2018. 43
See more
Pancreatic enzyme replacement therapy (PERT) is necessary to help a person with CF digest their food. This is because most people with CF have a condition known as pancreatic insufficiency. Enzymes come in capsule form as soon as the person with CF gets old enough to …
Who is the longest living person with cystic fibrosis?
Medication Digestive Treatments Chest Physical Therapy (CPT) Exercise Gene Therapy Clinical Trials Surgery There’s no cure for cystic fibrosis (CF). But many treatments can reduce your symptoms and...
What is routine care of patients with cystic fibrosis include?
Is cystic fibrosis caused by a dominant or recessive allele?
Is cystic fibrosis a Th17 disease?
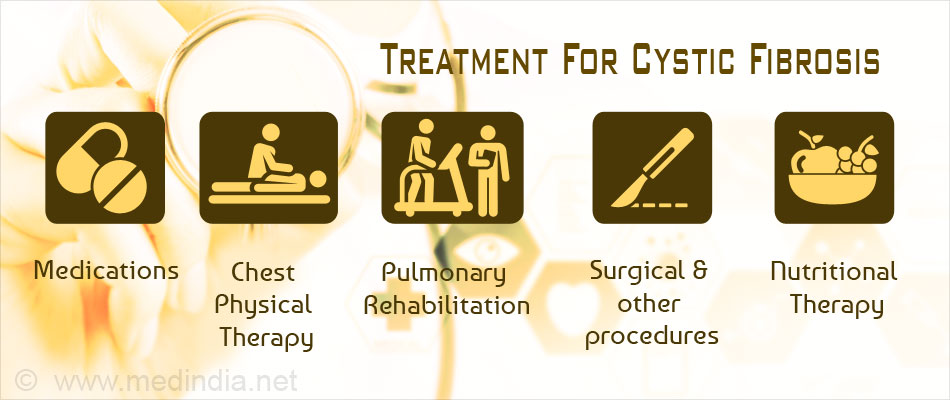
What are the best treatments for CF?
Medications. Options include: Medications that target gene mutations, including a new medication that combines three drugs to treat the most common genetic mutation causing CF and is considered a major achievement in treatment. Antibiotics to treat and prevent lung infections.
What tests are done for cystic fibrosis?
Doctors may also recommend genetic tests for specific defects on the gene responsible for cystic fibrosis. Genetic tests may be used in addition to checking the IRT levels to confirm the diagnosis.
Why does CF cause malnourishment?
Cystic fibrosis can cause malnourishment because the enzymes needed for digestion can't reach your small intestine, preventing food from being absorbed. People with CF may need a much higher number of calories daily than do people without the condition.
How to diagnose cystic fibrosis?
Diagnosis. To diagnose cystic fibrosis, doctors typically do a physical exam, review your symptoms and conduct several tests.
What is CTFR modulator?
For those with cystic fibrosis who have certain gene mutations, doctors may recommend cystic fibrosis transmembrane conductance regulator (CTFR) modulators. These newer medications help improve the function of the faulty CFTR protein. They may improve lung function and weight, and reduce the amount of salt in sweat.
How to test for cystic fibrosis in infants?
To evaluate if an infant has cystic fibrosis, doctors may also conduct a sweat test once the infant is at least 2 weeks old. A sweat-producing chemical is applied to a small area of skin. Then the sweat is collected to test it and see if it's saltier than normal.
Is there a cure for cystic fibrosis?
There is no cure for cystic fibrosis, but treatment can ease symptoms, reduce complications and improve quality of life. Close monitoring and early, aggressive intervention is recommended to slow the progression of CF, which can lead to a longer life.
Who treats cystic fibrosis?
This team will include pulmonologists, gastroenterologists, endocrinologists, nutritionists, nurses, and physical therapists.
What are the treatments for fibrosis?
Cystic fibrosis affects systems and organs throughout the body, so many other types of drugs will be relied on to treat symptoms, such as anti-inflammatory medications, prokinetics to treat gastroesophageal reflux, laxatives for intestinal obstruction, bile acids for liver blockage, and insulin for diabetes.
Why is mucus sticky in the lungs?
This is the basic mechanism responsible for cystic-fibrosis-related diseases throughout the body: Mucus in the lungs is too sticky to clear out and provides a rich environment for bacteria growth , resulting in chronic lung infections, bronchial damage (bronchiectasis), and scarring (fibrosis).
What causes mucus to be sticky?
Cystic fibrosis (or mu coviscidosis) is an inherited condition that reduces the water content of secretions within the body, causing thick and sticky mucus which fills up and blocks the lungs and other organs. It is one of the most common heritable genetic disorder in Caucasians in the US.
How many mutations are there in cystic fibrosis?
There are over 2000 genetic mutations that are responsible for cystic fibrosis. All are related to the CFTR protein. Based on the type of mutation, cystic fibrosis is divided into five classes of decreasing severity. Class 1 CF means cells are unable to produce working copies of CFTR proteins.
How to clear mucus from lungs?
One of the primary goals of cystic fibrosis treatment is to clear mucus from the lungs using physical therapy combined with mucus thinners taken through an inhaler or nebulizer. Mucolytics, such as dornase alfa, break up substances in the mucus, making it less sticky and easier to expel. Secretolytics, such as inhaled hypertonic saline solution, make mucus more watery by drawing water out of the tissues and into the airways. The effects of mucus thinners are temporary, so cystic fibrosis patients need to perform physical airway clearance to clear the thinned mucus.
Can a sweat test show CF?
A sweat test might show higher than normal salt levels, but not high enough for a CF diagnosis. A DNA test will then be used that looks for at least two copies of a cystic fibrosis mutation. There must be one copy on both chromosomes. The disease usually does not manifest if only one mutated gene is present.
What is the best treatment for cystic fibrosis?
bronchodilators to widen the airways and make breathing easier. steroid medicine to treat small growths inside the nose (nasal polyps) It's also important that people with cystic fibrosis are up-to-date with all routine vaccinations and have the flu jab each year once they're old enough.
How to clear mucus from lungs?
Exercise . Any kind of physical activity, like running, swimming or football, can help clear mucus from the lungs and improve physical strength and overall health. A physiotherapist can advise on the right exercises and activities for each individual.
How to avoid malnutrition?
A dietitian will advise on how to take in extra calories and nutrients to avoid malnutrition. They may recommend a high-calorie diet, vitamin and mineral supplements, and taking digestive enzyme capsules with food to help with digestion. The Cystic Fibrosis Trust has more information on nutrition and eating well.
What is an airway clearance device?
airway clearance devices – handheld devices that use breathing techniques, vibration and air pressure to help remove mucus from the airways (for example, a positive expiratory pressure, or PEP, device) The Cystic Fibrosis Trust has more information on airway clearance techniques and physiotherapy.
Can cystic fibrosis be treated with a lung transplant?
In severe cases of cystic fibrosis, when the lungs stop working properly and all medical treatments have failed to help, a lung transplant may be recommended. A lung transplant is a serious operation that carries risks, but it can greatly improve the length and quality of life for people with severe cystic fibrosis.
Is there a cure for cystic fibrosis?
There's no cure for cystic fibrosis, but a range of treatments can help control the symptoms, prevent or reduce complications, and make the condition easier to live with. Regular appointments to monitor the condition are needed and a care plan will be set up based on the person's needs.
What is CF in medical terms?
Cystic fibrosis (CF) is a hereditary, multisystemic disease caused by different mutations in the CFTRgene encoding CF transmembrane conductance regulator. CF is mainly characterized by pulmonary dysfunction as a result of deterioration in the mucociliary clearance and anion transport of airways. Mortality is mostly caused by bronchiectasis, ...
What is CFTR in the body?
CFTR acts as a cAMP regulated chlorine channel in apical membranes, providing Na+and water transport from epithelial cells in many organs and glands.4CFTR dysfunction primarily affects epithelial cells and causes chronic microbial infection and subsequently airway inflammation. Mortality from CF is commonly caused by bronchiectasis, ...
Where is CFTR located?
CF is caused by different mutations in the CFTR gene encoding CF transmembrane conductance regulator (CFTR), which regulates the mucociliary clearance and anion transport in airways.3The CFTR gene is located on the long arm of chromosome 7 and the CFTR protein product is 1,480 amino acids in length. CFTR acts as a cAMP regulated chlorine channel in ...
Is CFTR a heterogeneous disease?
Organoids. As CF is a genetically heterogeneous disease, currently available treatment options do not cover all CFTR mutations. Many of the known CFTR mutations are associated with a variety of disease expression and this complicates the estimation of individual disease phenotypes.
What is the first CFTR potentiator?
The first small molecule defined as a CFTR potentiator (potential enhancer) is ivacaftor, which was developed as VX-770 at first.69Ivacaftor facilitates the transport of chloride by enhancing the channel opening of the CFTR protein on the cell surface.
What is a tezacaftor?
Tezacaftor (VX-661) enhances the processing and transfer of CFTR proteins, including both normal and mutant ones (including ΔF508-CFTR), and thus increases the amount of protein reaching the cell surface. The tezacaftor/ivacaftor combination was approved by the FDA in 2018.
Is CF a hereditary disease?
Cystic fibrosis (CF) is a hereditary, multifactorial, multisystemic disease characterized by obstruction of airways, microbial infection, digestive disorders, and other complications. CF is known as the most common autosomal recessive disease in Caucasians.1. Although the incidence of disease varies greatly throughout the world, ...
How to help cystic fibrosis patients?
That’s why it’s important to find ways to manage stress. Yoga, meditation, exercise, hanging out with friends, and hobbies are all popular and effective ways to manage stress, anxiety, ...
How to help CF?
Exercise. Among its many benefits for people with CF, exercise builds lung capacity, helps with airway clearance, builds strong bones, and strengthens the heart and breathing muscles. Dedication to daily exercise keeps people with CF healthier, longer.
How far away should you be from someone with cystic fibrosis?
The 6-foot rule. People with cystic fibrosis are especially vulnerable to lung infections carried by other people with CF. That is why it’s important for people with CF to keep a distance of 6 feet from others with CF from a different household.
Why do people with CF need extra calories?
But these can’t just be empty calories. Due to their gastrointestinal system not digesting food properly, a condition called malabsorption, people with CF need extra nutrition to prevent malnutrition and failure to thrive as children. 3
How much time do you spend on CF?
It varies by individual, but the average amount of time an adult with CF spends on their treatments (medications, enzymes, and airway clearance) is 108 minutes per day, regardless of the severity of their disease. Caregivers of children with CF report 74 minutes ...
What is airway clearance?
Airway clearance helps loosen the thick, sticky mucus that tends to clog the lungs of people with CF. The type of airway clearance technique (ACT) used varies by age and which method the person with CF prefers. Parents must perform ACTs for infants and toddlers, while older children and adults perform their own.
What to do if you have CF?
These might include: Sinus surgery: Many people with CF have inflamed or infected sinuses. Your doctor may need to remove nasal polyps (growths inside your nasal passages). They can also do a procedure called an “ endoscopy and lavage” that suctions mucus from your airways. This will make it easier for you to breathe.
How to help a lung infection?
They may also help reduce the number of lung infections you get. For instance, clapping or pounding on your chest and back helps loosen mucus so you can cough more of it out. You can do different types of ACTs at home with the help of a family member or friend. Or, you might prefer to use a medical device.
Is there a cure for cystic fibrosis?
There’s no cure for cystic fibrosis (CF). But many treatments can reduce your symptoms and improve your quality of life. Here’s an overview of the most common.
Diagnosis
Treatment
Clinical Trials
Coping and Support
Specialist to consult
Preparing For Your Appointment