
Medication
NICHD-supported researchers and other scientists are exploring additional treatments for PKU. These treatments include large neutral amino acid supplementation, which may help prevent phenylalanine from entering the brain, and enzyme replacement therapy, which uses a substance similar to the enzyme that usually breaks down phenylalanine.
Self-care
Phenylketonuria (PKU) is a rare hereditary condition in which the amino acid phenylalanine is not properly metabolized. PKU can cause severe mental retardation if not treated. The following list of medications are in some way related to, or used in the treatment of this condition.
Nutrition
The diet should not be terminated after adolescence, because strong evidence indicates that hyperphenylalaninemia can have detrimental effects in adult patients. Some adults with untreated PKU who have cognitive decline may show improvement in behavior and physical manifestations when treated with a phenylalanine-restricted diet.
What are the treatment options for phenylketonuria?
Newborn blood testing identifies almost all cases of phenylketonuria. All 50 states in the United States require newborns to be screened for PKU. Many other countries also routinely screen infants for PKU.
What is phenylketonuria?
What is the prognosis of phenylalanine-restricted diet for phenylketonuria (PKU)?
What tests are used to diagnose phenylketonuria (PKU)?
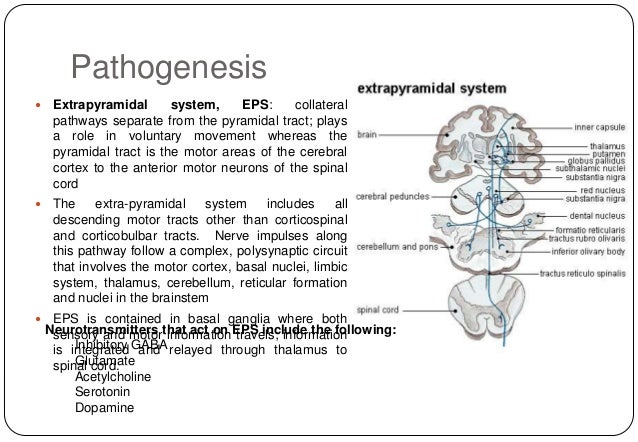
What is the main treatment for phenylketonuria?
The main treatment for PKU is a low-protein diet that completely avoids high-protein foods (such as meat, eggs and dairy products) and controls the intake of many other foods, such as potatoes and cereals.
Can phenylketonuria be treated?
There is no cure for PKU. The most important treatment is a diet that limits foods with phenylalanine. This means the diet must be low in protein. Newborns diagnosed with the disease must use special infant formula.
Which amino acid is needed to be avoided in treatment of phenylketonuria?
Phenylketonuria (PKU) is caused by deficient activity of the enzyme phenylalanine hydroxylase, needed to convert the essential amino acid (AA) phenylalanine (phe) to tyrosine.
Can phenylketonuria be treated with gene therapy?
There is an important need to develop therapies for PKU that will correct behavioral abnormalities in adults to improve their quality of life. Successful gene therapy for PKU in adults will enable treatment for infants to prevent early damage due to Phe accumulation and provide a life-long cure.
What medicines contain phenylalanine?
Drug Products Containing PhenylalaninePRODUCTPHE CONTENTAugmentin 400mg4.2mg/tabletBenadryl allergy and sinus fastmelt4.6mg/tabletBenadryl allergy chewables4.2mg/tabletBenadryl, Childrens allergy and cold fast melt tablets4.6mg/tablet137 more rows
How does tyrosine work?
Tyrosine is in all tissues of the human body and in most of its fluids. It helps the body build proteins in your body, and produce enzymes, thyroid hormones, and the skin pigment melanin. It also helps the body produce neurotransmitters that help nerve cells communicate.
Which enzyme is essential for conversion of phenylalanine into tyrosine in PKU?
enzyme phenylalanine hydroxylaseInfants with PKU lack the ability to produce the enzyme phenylalanine hydroxylase, which converts phenylalanine to tyrosine.
Is phenylalanine an essential amino acid?
Essential amino acids cannot be made by the body. As a result, they must come from food. The 9 essential amino acids are: histidine, isoleucine, leucine, lysine, methionine, phenylalanine, threonine, tryptophan, and valine.
How do you reduce phenylalanine?
Controlling blood phenylalanine during pregnancy Eat regular small, frequent low phenylalanine meals. Low protein special foods are an important source of calories and should be incorporated into the diet each day. Weight loss can lead to higher blood phenylalanine levels.
Is PKU a good candidate for gene therapy?
Since PKU is probably a candidate for gene therapy in vivo, a significant improvement of the delivery system and persistent gene expression will be required before clinical application can be considered.
What is the PAH gene?
The PAH gene provides instructions for making an enzyme called phenylalanine hydroxylase. This enzyme is responsible for the first step in processing phenylalanine, which is a building block of proteins (an amino acid) obtained through the diet. Phenylalanine is found in all proteins and in some artificial sweeteners.
What is gene therapy simple definition?
Gene therapy is a technique that modifies a person's genes to treat or cure disease. Gene therapies can work by several mechanisms: Replacing a disease-causing gene with a healthy copy of the gene. Inactivating a disease-causing gene that is not functioning properly.
How to diagnose phenylketonuria?
Phenylketonuria is generally diagnosed through newborn screening. Once your child is diagnosed with PKU, you'll likely be referred to a medical center or specialty clinic with a doctor who specializes in treating PKU and a dietitian with expertise in the PKU diet.
Why do we need a PKU formula?
Taking a PKU formula — a special nutritional supplement — for life to make sure you get enough essential protein (without phenylalanine) and nutrients that are crucial for growth and general health.
What is the National PKU Alliance?
The National PKU Alliance is an online support group for adults with PKU. Get help with menu planning. A registered dietitian with experience in PKU can help you devise delicious low-phenylalanine dinners. He or she may also have great ideas for holiday meals and birthdays. Plan ahead when you eat out.
What is the best supplement for PKU?
Neutral amino acid therapy. Another possible addition to the PKU diet is a supplement called neutral amino acid therapy in powder or tablet form. This supplement may block some absorption of phenylalanine. This may be a treatment option for adults with PKU.
How long after birth can you test for PKU?
Testing your baby after birth. A PKU test is done a day or two after your baby's birth. The test is done after your baby is 24 hours old and after your baby has ingested some protein in the diet to ensure accurate results. A nurse or lab technician collects a few drops of blood from your baby's heel or the bend in your baby's arm.
What to eat for PKU?
Plan ahead, so there's always a PKU-friendly food option. Pack dehydrated fruit snacks, raisins and lower protein crackers for the car. Take fruit kebabs or vegetable skewers to a cookout, and make a low-phenylalanine salad for the neighborhood potluck.
Can a baby with PKU eat formula?
Because regular infant formula and breast milk contain phenylalanine, babies with PKU instead need to consume a phenylalanine-free infant formula. A dietitian can carefully calculate the amount of breast milk or regular formula to be added to the phenylalanine- free formula.
Drugs used to treat Phenylketonuria
The following list of medications are in some way related to, or used in the treatment of this condition.
Further information
Always consult your healthcare provider to ensure the information displayed on this page applies to your personal circumstances.
What causes phenylketonuria?
Causes of phenylketonuria. PKU is an inherited condition caused by a defect in the PAH gene. The PAH gene helps create phenylalanine hydroxylase, the enzyme responsible for breaking down phenylalanine. A dangerous buildup of phenylalanine can occur when someone eats high-protein foods, such as eggs and meat.
How severe is phenylketonuria?
Symptoms of phenylketonuria. PKU symptoms can range from mild to severe. The most severe form of this disorder is known as classic PKU. An infant with classic PKU may appear normal for the first few months of their life.
How to test for PKU in newborn?
Since the 1960s, hospitals in the United States have routinely screened newborns for PKU by taking a blood sample. A doctor uses a needle or lancet to take a few drops of blood from your baby’s heel to test for PKU and other genetic disorders.
How many babies are diagnosed with PKU?
Babies in the United States are screened for PKU shortly after birth. The condition is uncommon in this country, only affecting about 1 in 10,000 to 15,000 newborns each year. The severe signs and symptoms of PKU are rare in the United States, as early screening allows treatment to begin soon after birth. Early diagnosis and treatment can help ...
Why do babies get PKU?
PKU is caused by a defect in the gene that helps create phenylalanine hydroxylase. When this enzyme is missing, your body can’t break down phenylalanine. This causes a buildup of phenylalanine in your body. Babies in the United States are screened for PKU shortly after birth.
What is the name of the condition where a protein is made from an amino acid called?
Phenylketonuria (PKU) is a rare genetic condition that causes an amino acid called phenylalanine to build up in the body. Amino acids are the building blocks of protein. Phenylalanine is found in all proteins and some artificial sweeteners. Phenylalanine hydroxylase is an enzyme your body uses to convert phenylalanine into tyrosine, ...
What is a PKU?
A less severe form of PKU is called variant PKU or non-PKU hyperphenylalaninemia. This occurs when the baby has too much phenylalanine in their body. Infants with this form of the disorder may have only mild symptoms, but they’ll need to follow a special diet to prevent intellectual disabilities.
How to diagnose PKU?
Diagnosing PKU. Your baby will have been tested a day or two after birth. This test is done with a heel stick, and a small amount of blood is collected for this and other routine tests. If the test indicates that your baby may have this disorder, additional test will be performed.
What are the different types of PKU?
Types of PKU. There are four types of PKU: Hyperphenylalaninemia: the lowest level above normal. Mild PKU: blood levels are mildly elevated. Moderate or variant: levels are not low but not high. Classic PKU: blood levels of phenyla nine are high.
What is PKU in biology?
What is Phenylketonuria (PKU)? Phenylketonuria is an inborn error of protein metabolism. It is a rare disease, and children who are born with this condition inherit it from their parents. This condition prevents the body from being able to properly break down proteins — specifically phenylanine, which is found in protein.
What happens if a pregnant woman has PKU?
Seizures. If a pregnant mother has PKU that is not controlled by diet, her baby may have a more severe form of this disorder. This can cause birth defects in the baby, such as a small head (microcephaly) and heart problems.
How do you know if you have a PKU?
Unless a child is born with birth defects, symptoms of PKU may not become noticeable for a few months. These symptoms in young babies can include: Eczema, a skin rash. Seizures. Slow growth. A musty body odor or breath. Uncontrolled PKU can lead to other problems as the child grows, such as: Developmental delays.
Why is phenylanine bad for you?
When the body is unable to break down certain proteins, phenylanine builds up in the blood and causes problems throughout the body. Some ways that too much phenylanine can affect the body are: If a pregnant mother has PKU that is not controlled by diet, her baby may have a more severe form of this disorder.
What is the goal of phenylanine treatment?
If your child has this disorder, he or she will need to begin treatment as soon as possible. The goal of treatment is to keep blood levels of phenylanine low.
What is PKU in biology?
Phenylketonuria (PKU) is an autosomal recessive inborn error of metabolism caused by a deficiency in the hepatic enzyme phenylalanine hydroxylase (PAH). If left untreated, the main clinical feature is intellectual disability.
What foods can you eat with PKU?
Patients with PKU must strictly limit their intake of foods rich in protein, such as meats, fish, eggs and dairy products. Low-protein high-starch natural foods such as potatoes, some vegetables (such as peas) can be eaten but only in restricted amounts.
What enzyme converts phe to tyrosine?
PAH is a hepatic enzyme that requires the cofactor tetrahydrobiopterin (BH4) to convert Phe to tyrosine (Tyr) (Figure 1). A deficiency in PAH or its cofactor BH4, results in the accumulation of excess phenylalanine, whose toxic effects can cause severe and irreversible intellectual disability if untreated (1).
Which country has the lowest prevalence of PKU?
Finland has the lowest incidence in Europe with one case in every 100,000 live births, while Turkey has the highest incidence with one in every 4000 births due to high consanguinity within the population (5). In Australia, approximately 25 babies are diagnosed with PKU each year (based on the recorded incidence of new cases).
Is PKU a maternal condition?
Furthermore, females with PKU are at high risk of having a child with the so-called maternal PKU syndrome, causing microcephaly, intra-uterine growth restriction, congenital heart defects, a characteristic facial appearance and cognitive impairment in the affected infant (29,30).
What is the treatment for PKU?
Treatment consists of dietary restriction of phenylalanine often with tyrosine supplementation.
What is PKU in psychology?
Most patients with phenylketonuria (PKU) are treated in a specialty metabolic disease clinic, and such patients are probably best served by being followed in such a clinic. A psychologist should perform developmental testing at regular intervals. Whenever possible, the patient and parents should work with a nutritionist experienced in PKU, usually as part of a PKU or metabolic disease clinic.
How much phenylalanine is in a 12 oz can of aspartame?
A 12-oz can of aspartame-sweetened diet drink contains approximately 105 mg of phenylalanine (ie, 25-50% of the usual daily intake). Most newborns with PKU require 40 mg/kg/d to 60 mg/kg/d of dietary phenylalanine to maintain normal growth and development.
How often should phenylalanine be monitored?
Phenylalanine levels are monitored typically twice a week in neonates, weekly in infants, biweekly or every 3 weeks in toddlers, and monthly thereafter, even during adult life. Attention should be given to variability in blood phenylalanine levels and to maintenance within the recommended range.
Why is phenylalanine important?
Providing some natural phenylalanine is essential in order to prevent deficiency of this essential amino acid. The diet requires virtual elimination of all high-protein foods, such as meat, dairy, nuts, and legumes.
What are some supplements that are low in protein?
Supplements including capsules, amino acid bars, and amino acids cooked into foods, are becoming available. Energy and variety are provided by low-protein foods, including fruits and nonstarchy vegetables , as well as specially ordered low-protein foods.
Can you treat PKU before conception?
Treatment at any time during pregnancy may reduce the severity of developmental delay. Women with PKU should start a phenylalanine-restricted diet before conception, and those contemplating pregnancy or who are pregnant should be treated in metabolic or PKU clinics. Previous.
Diagnosis
Treatment
Lifestyle and Home Remedies
Coping and Support

Specialist to consult
Preparing For Your Appointment
Causes
- Starting treatment early and continuing treatment throughout life can help prevent intellectual disability and major health problems. The main treatments for PKUinclude: 1. A lifetime diet with very limited intake of foods with phenylalanine 2. Taking a PKUformula — a special nutritional supplement — for life to make sure that you get enough essent...
Epidemiology
- Strategies to help manage PKUinclude keeping track of foods eaten, measuring correctly, and being creative. Like anything, the more these strategies are practiced, the greater the comfort and confidence you can develop.
Symptoms
- Living with PKUcan be challenging. These strategies may help: 1. Stay informed. Knowing the facts about PKU can help you take charge of the situation. Discuss any questions with your pediatrician, family health care provider, geneticist or dietitian. Read books and cookbooks specifically written for people with PKU. 2. Learn from other families. Ask your health care provi…
Variations
- Phenylketonuria is generally diagnosed through newborn screening. Once your child is diagnosed with PKU, you'll likely be referred to a medical center or specialty clinic with a specialist who treats PKU and a dietitian with expertise in the PKUdiet. Here's some information to help you get ready for your appointment and know what to expect.
Prognosis
Pathophysiology
Genetics
Diagnosis
Preparation
Treatment
Diet
Medical uses
Risks
Prevention