Can NMDA receptor antagonists help fight Alzheimer's?
But doctors do use one NMDA receptor antagonist: memantine ( Namenda XR). It's been approved in the U.S. and Europe as a treatment for Alzheimer's disease. …
What is NMDA antidepressant?
Jul 12, 2020 · Memantine, a partial antagonist of N-methyl-D-aspartate receptor (NMDAR), approved for moderate to severe Alzheimer's disease (AD) treatment within the U.S. and Europe under brand name Namenda (Forest), Axura and Akatinol (Merz), and Ebixa and Abixa (Lundbeck), may have potential in alleviating additional neurological conditions, such as vascular dementia …
What is N-methyl D-aspartate (NMDA) antagonist?
Feb 08, 2019 · Memantine, a non-competitive NMDA receptor antagonist, is approved for use in moderate to severe AD. It has been widely prescribed to provide symptomatic relief and enhance life quality in AD, even if it did not improve excessive agitation (Fox et al., 2012), and hippocampal or total brain atrophy (Wilkinson et al., 2012). However, in the brain areas mainly affected in AD, …
Which subunits of the NMDA receptor are critical for long-term potentiation and depression?
Abstract. Research into Alzheimer's disease (AD) pathology has identified several underlying disease processes that are potential targets for drug discovery and development. One strategy targets glutamatergic neurotransmission mediated by the N-methyl-D-aspartate (NMDA) receptor. Therapeutic intervention with high-affinity NMDA receptor antagonists, such as …
What is the treatment for moderate to severe Alzheimer disease?
Which drug is indicated for use in mild moderate and severe Alzheimer's disease?
Which glutamate antagonist is used to treat Alzheimer's disease?
Which drug is effective as NMDA receptor antagonist?
What is donepezil used for?
What is memantine used for?
Is NMDA a glutamate?

Why is NMDA an antagonist for Alzheimer's?
What is the difference between AMPA and NMDA receptors?
Is glutamate an NMDA antagonist?
Is Tramadol an NMDA antagonist?
What does N-methyl-D-aspartate do?
What is the name of the drug that is used to treat Alzheimer's disease?
Memantine, a partial antagonist of N-methyl-D-aspartate receptor (NMDAR), approved for moderate to severe Alzheimer's disease (AD) treatment within the U.S. and Europe under brand name Namenda (Forest), Axura and Akatinol (Merz), and Ebixa and Abixa (Lundbeck), may have potential in alleviating addi …. N-methyl D-aspartate (NMDA) ...
What is the N-methyl D-aspartate receptor antagonist?
N-methyl D-aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer's disease, vascular dementia and Parkinson's disease. Memantine, a partial antagonist of N-methyl-D-aspartate receptor (NMDAR), approved for moderate to severe Alzheimer's disease (AD) treatment within the U.S.
What is the therapeutic action of memantine?
The key to memantine's therapeutic action lies in its uncompetitive binding to the NMDAR through which low affinity and rapid off-rate kinetics of memantine at the level of the NMDAR-channel preserves the physiological function of the receptor, underpinning memantine's tolerability and low adverse event profile.
Does memantine block glutamate?
While excessive levels of glutamate result in neurotoxicity, in part through the over-activation of NMDARs, memantine-as a partial NMDAR antagonist, blocks the NMDA glutamate receptors to normalize the glutamatergic system and ameliorate cognitive and memory deficits.
What is the function of NMDARs?
NMDARs are permeable to Na+, K+, and high permeability to Ca2+, which acts as a second messenger to modify synapses (Lynch et al., 1983). NMDARs are essential mediators of brain plasticity and are capable of converting specific patterns of neuronal activity into long-term changes in synapse structure and function that are thought to underlie higher cognitive functions (Traynelis et al., 2010). Activation of NMDARs leads to cytosolic free intracellular calcium ([Ca2+]i) increase (MacDermott et al., 1986), required for long-term potentiation (LTP) and long-term depression (LTD) (Muller et al., 2009), and, more generally, for synaptic plasticity (MacDonald et al., 2006; Lau et al., 2009). NMDARs are also thought to be involved in a process called excitotoxicity. Abnormal NMDAR activity is associated with seizure, ischemic stroke (Choi et al., 1988; Villmann and Becker, 2007), and neurodegenerative disorders (Benarroch, 2011), such as Alzheimer’s (Wenk, 2006), Huntington’s (Fan and Raymond, 2007), and Parkinson’s disease (Bonuccelli and Del Dotto, 2006).
What are the subunits of NMDAR?
GluN2A and GluN2B are the major subunits of functional NMDARs. Some studies have suggested that GluN2A and GluN2B may differentially mediate NMDAR function at synaptic and extrasynaptic locations and play opposing roles in excitotoxicity (Thomas et al., 2006; Harris and Pettit, 2007). In the postsynaptic membrane of excitatory neurons, the density of NMDARs is higher in dendritic spines, within the postsynaptic density (PSD), considered synaptic NMDARs, than in the dendritic shaft and somatic membrane (Köhr, 2006). At the PSD, the receptors form a large macromolecular NMDAR complex (NRC), containing a vast collection of scaffolding, adaptor and effector proteins that are involved in activation of downstream signaling cascades and regulation of NMDAR function, membrane stability and trafficking (Sheng and Lee, 2000). Extrasynaptic NMDARs are localized at sites further from the PSD, on the spine neck, the dendritic shaft or soma (Newpher and Ehlers, 2008). Perisynaptic NMDARs are located on the plasma membrane (PM) within 200–300 nm of the PSD. The perisynaptic region may contain mobile receptors that are in transit to and from the PSD (Zhang and Diamond, 2006; Petralia et al., 2009). Synaptic and extrasynaptic NMDARs are gated by different endogenous co-agonists: D-serine for synaptic NMDARs and glycine for extrasynaptic NMDARs. Both co-agonists are of glial origin (Papouin et al., 2012). It was proposed that GluN2A- and GluN2B-containing receptors are predominantly found synaptically and extrasynaptically, respectively. For NMDAR redistribution plays a larger role during development, when there is a synaptic switch from GluN2B to GluN2A-containing receptors (Barria and Malinow, 2002). In addition, it has been found that synaptic and extrasynaptic NMDA receptors have opposing effects in determining the fate of neurons (Vizi et al., 2013). Calcium entry through synaptic GluN2A-containing NMDARs induces activity of cAMP response element binding protein (CREB) and gene expression of CREB-evoked brain-derived neurotrophic factor (BDNF), which is involved in the neuroprotective action of synaptic glutamatergic transmission. In contrast, Ca2+entry through GluN2B-containing NMDARs, which are expressed extrasynaptically, triggers the CREB shut-off pathway (Leveille et al., 2008). There is some evidence that synaptic NMDARs support LTP, while extrasynaptic NMDARs mediate LTD in the mature brain (Massey, 2004). Specific NMDAR subunits are not confined to particular subcellular locations on PM. Some evidences shown that GluN2A- and GluN2B-subtypes are present at both synaptic and extrasynaptic sites (Thomas et al., 2006; Harris and Pettit, 2007; Martel et al., 2009; Zhou et al., 2013a). These suggest that any difference in signaling by synaptic and extrasynaptic NMDARs must be due to intracellular signaling pathways rather than subunit identity or mobility (Harris and Pettit, 2007). And some researchers thought that these results were partially refuted on the basis that these experiments were carried out on developing hippocampal cultured neurons; the same results may not be found under in vivoconditions (Newpher and Ehlers, 2008).
What proteins are involved in synaptic plasticity?
Proteins interacting with NMDAR subunits are therefore important determinates for the direction of synaptic plasticity. The differential interaction of GluN2A and GluN2B subunits to membrane-associated guanylate kinase (MAGUKs) is controversial. It was once believed that GluN2A preferentially bound to postsynaptic density protein-95 (PSD-95), while GluN2B preferentially bound synapse-associated protein 102 (SAP102) (Sans et al., 2000; Krapivinsky et al., 2003). GluN2B interacts directly with Ras-guanine nucleotide releasing factor 1 (Ras-GRF1) (Krapivinsky et al., 2003), and synaptic Ras GTPase activating protein (RasGAP), presumably through SAP102 (Kim et al., 1998). The unique associations are likely to affect the induction of plasticity (Zhu et al., 2002). Moreover, these interactions were thought to control distinct synaptic localization of GluN2A and GluN2B (Townsend et al., 2003). However, a recent biochemical study using a serial immunoprecipitation suggests that MAGUK proteins such as PSD-95 and SAP102 interact with diheteromeric GluN1/GluN2A and GluN1/GluN2B receptors at comparable levels (Al-Hallaq et al., 2007). Additional studies are needed to clarify the association of NMDAR subunits with MAGUK family members and what effects these associations may have on receptor localization and on plasticity signaling pathways (Yashiro and Philpot, 2008).
What are the two tetrameric receptors?
These tetrameric receptors consist of two obligatory GluN1 subunits and two regulatory subunits, usually a combination of GluN2A and GluN2B. Among the six regulatory subunits of NMDARs, GluN2A and GluN2B have been the most extensively studied because they are broadly expressed in the brain, predominate in the postnatal cortex, and are believed to play important roles in synaptic plasticity. In the neonatal neocortex GluN2B-containing NMDARs predominate, and sensory experience facilitates a developmental switch in which GluN2A levels increase relative to GluN2B (Yashiro and Philpot, 2008). However, GluN1/GluN2B carry about twofold more charge for a single synaptic event than GluN1/GluN2A channels (Erreger et al., 2005). More studies showed that GluN2A-dominated synapses are more likely to induce LTD than GluN2B-dominated synapses, while GluN2B-dominated synapses have a greater capacity to be potentiated. GluN2A co-immunoprecipitates with neuronal nitric oxide (NO) synthase more effectively than GluN2B (Al-Hallaq et al., 2007). Although this interaction is likely indirect, the association raises the interesting possibility that NO-mediated presynaptic forms of LTP and LTD may be preferentially linked to GluN2A-mediated signaling pathways (Haghikia et al., 2007). Activated CaMKII binds strongly to GluN2B, which is required for LTP induction (Barria and Malinow, 2005). However, both GluN2A-containing and GluN2B-containing NMDARs are capable of supporting bidirectional synaptic plasticity (Yashiro and Philpot, 2008). GluN2A and GluN2B are present as either diheteromers (GluN1/GluN2A or GluN1/GluN2B) or triheteromers (GluN1/GluN2A/GluN2B) (Yashiro and Philpot, 2008). In isolated GluN2A-only or GluN2B-only synapses, GluN1/GluN2A diheteromeric channels exhibit faster rising and decaying currents than GluN1/GluN2B diheteromeric channels (Prybylowski et al., 2002). GluN1/GluN2A/GluN2B triheteromeric channels appear to exhibit intermediate decay time courses between the two diheteromeric channel types (Vicini et al., 1998).
What are the functions of GluN2A and GluN2B?
In the adult CNS, particularly in higher brain structures, such as the hippocampus and cortex, GluN2A and GluN2B are the predominant subunits, indicating that they have central roles in synaptic function and plasticity (Monyer et al., 1994; Takai et al., 2003). GluN2C- or GluN2D- containing receptors appear to give rise to ‘low-conductance’ openings with a lower sensitivity to extracellular Mg2+, which may affect the Ca2+influx generated by synaptic activation of NMDAR (Momiyama et al., 1996). Misra et al. (2000)results indicate that GluN1/GluN2D receptors do not contribute to the excitatory postsynaptic current (EPSC) and appear to be restricted to the extrasynaptic membrane. The possibility still exists that the GluN2D subunit is present at the synapse but is preferentially co-assembled with other GluN2 subunits, such as triheteromeric assemblies (GluN1/GluN2B/GluN2D) (Dunah et al., 1998; Cull-Candy et al., 2001). GluN3A and GluN3B were cloned based on similarity to GluN1 and GluN2 subunits and were the last NMDA receptor subunits to be discovered (Low and Wee, 2010). GluN3A expression is low embryonically, peaks during early postnatal life, and diminishes to lower levels in adulthood. This expression profile is observed in many regions of the brain (Bendová et al., 2009; Roberts et al., 2009). Conversely, GluN3B levels are low around postnatal day 10 and gradually increase into adulthood within the neocortex, hippocampus, striatum, cerebellum, brainstem, and spinal cord (Wee et al., 2008). GluN3A and GluN3B are expressed by multiple neuronal cell types, including interneurons, pyramidal cells, motor neurons, trigeminal neurons, retinal ganglion and amacrine cells (Pachernegg et al., 2012). Although GluN3A is present in oligodendrocytes, it does not seem to be expressed in astrocytes (Matsuda et al., 2003; Káradóttir et al., 2005; Salter and Fern, 2005). The GluN3 subunits bind glycine and D-serine (Yao, 2006; Yao et al., 2008), but the functional properties and physiological roles of GluN3-containing NMDA receptors remain elusive. GluN3A acts in a dominant-negative manner to suppress receptor activity. GluN3A containing NMDARs display striking regional and temporal expression specificity, and, unlike most other NMDAR subtypes, they have a low conductance, are only modestly permeable to Ca2+, and pass current at hyperpolarized potentials in the presence of magnesium. While glutamate activates triheteromeric NMDARs composed of GluN1/GluN2/GluN3A subunits, glycine is sufficient to activate diheteromeric GluN1/GluN3A-containing receptors (Henson et al., 2010). About GluN3A is expressed in the right place at the right time to regulate spine and synapse development, there are two hypotheses, if the influence of GluN3A is to serve as a ‘synaptic brake’ to limit synapse/spine formation or if it serves as a ‘synaptic marker’ to promote the elimination of spines (Henson et al., 2010). Roberts et al. (2009)found that prolonging GluN3A expression prevents glutamatergic synapse maturation by limiting synapse potentiation and growth, and decreasing spine density, accompanied by major impairments in learning and memory processes. Whereas knocking out endogenous GluN3A conversely accelerates synaptic maturation events.
How many NMDAR subunits are there?
Seven different NMDAR subunits have been identified: the GluN1 subunit, four distinct GluN2 subunits (GluN2A, GluN2B, GluN2C, and GluN2D), and a pair of GluN3 subunits (GluN3A and GluN3B) (Paoletti, 2011). Functional NMDARs are heterotetramers composed of two glycine or D-serine-binding GluN1 subunits and two glutamate-binding GluN2 (GluN2A-D) subunits or, in some cases, glycine-binding GluN3 (GluN3A/B) subunits (Köhr, 2006). All NMDAR subunits share a common membrane topology that contain four discrete domains (Lee et al., 2014; Karakas and Furukawa, 2014): The extracellular amino-terminal domain (ATD) is involved in subunit assembly and allosteric regulation, and contributes to control of ion channel open probability and deactivation speeds. The extracellular ligand-binding domain (LBD) is formed by two discontinuous segments (S1 and S2), and binds agonists and antagonists to control ion channel opening. The transmembrane domain (TMD) contains three transmembrane segments (M1, M3, and M4) and a re-entrant pore loop (M2) which is part of the channel pore, containing a critical asparagine residue that determines calcium permeability of the channel and mediates the magnesium blockade (Paoletti et al., 2013; Karakas and Furukawa, 2014). The intracellular carboxyl-terminal domain (CTD) interacts with multiple cytosolic proteins (Sanz-Clemente et al., 2012). GluN2 LBDs contain the glutamate binding site, while GluN1 (or GluN3) LBDs contain the Glycine/D-serine coagonist binding site (Hackos and Hanson, 2017). In general, activation of NMDARs containing GluN1/GluN2 requires binding of both glycine and glutamate at the extracellular LBDs in addition to release of magnesium block by membrane depolarization at the TMD (Regan et al., 2015). Positively charged residues on the surface of the GluN2A LBD assist glutamate binding via a ‘guided-diffusion’ mechanism. On the other hand, Glycine binds to the GluN1 LBD via an ‘unguided-diffusion’ mechanism, whereby glycine finds its binding site primarily by random thermal fluctuations (Yu and Lau, 2018). A recent study show, through cryoelectron microscopy, that the presence of the exon 5 motif, at the ATD of the GluN1 subunit, alters the local architecture of heterotetrameric GluN1/GluN2 NMDA receptors and creates contacts with the LBDs of the GluN1 and GluN2 subunits. The unique interactions established by the exon 5 motif are essential to the stability of the ATD/LBD and LBD/LBD interfaces that are critically involved in controlling proton sensitivity and deactivation (Regan et al., 2018). The most widely expressed NMDARs contain the obligatory subunit GluN1 plus either GluN2B or GluN2A or a mixture of the two; therefore, in next sections we will focus on these two subunits.
Does glutamate activate extrasynaptic NMDARs?
Extrasynaptic NMDARs are exposed to ambient glutamate, whether this glutamate concentration is high enough to tonically activate extrasynaptic NMDARs remains controversial. Although microdialysis studies report that ambient glutamate concentrations in vivoare high enough to activate extrasynaptic NMDARs (Nyitrai et al., 2006), a study suggests that glutamate transporters regulate ambient glutamate concentrations at a level that is too low to cause significant receptor activation (Herman and Jahr, 2007). While, some reports that glutamate that is released into the extracellular space mainly from glial processes (Fellin et al., 2004) may result in the persistent activation of extrasynaptic GluN2B receptors, which are of high affinity and are sensitive to low concentrations of glutamate (Vizi, 2000).
What is the drug used to treat Alzheimer's?
A medication known as memantine, an N-methyl D-aspartate (NMDA) antagonist, is prescribed to treat moderate to severe Alzheimer’s disease. This drug’s main effect is to decrease symptoms, which could enable some people to maintain certain daily functions a little longer than they would without the medication.
What is the FDA's new drug for Alzheimer's?
And, on June 7, 2021, FDA provided accelerated approval for the newest medication, aducanumab, which helps to reduce amyloid deposits in the brain and may help slow the progression of Alzheimer’s, although it has not yet been shown to affect clinical symptoms or outcomes, such as progression of cognitive decline or dementia.
What is the best treatment for Alzheimer's?
Aducanumab is the only disease-modifying medication currently approved to treat Alzheimer’s. This medication is a human antibody, or immunotherapy, that targets the protein beta-amyloid and helps to reduce amyloid plaques, which are brain lesions associated with Alzheimer’s.
How is aducanumab approved?
Aducanumab was approved through the FDA’s Accelerated Approval Program, which provides a path for earlier approval of drugs that treat certain serious conditions. This helps people living with the disease gain earlier access to the treatment. The approval of aducanumab was based on the ability of the drug to reduce amyloid in the brain. When using the accelerated approval pathway, drug companies are required to conduct additional studies to determine whether there is in fact clinical benefit after the drug is approved. If the follow-up trial fails to verify clinical benefit, the FDA may withdraw approval of the drug. Results of the phase 4 clinical trial for aducanumab are expected to be available by early 2030.
How does memantine help Alzheimer's patients?
For example, memantine may help a person in the later stages of the disease maintain his or her ability to use the bathroom independently for several more months, a benefit for both the person with Alzheimer's and caregivers. Memantine is believed to work by regulating glutamate, an important brain chemical.
What is the National Institute on Aging's ADEAR Center?
The National Institute on Aging’s ADEAR Center offers information and free print publications about Alzheimer’s disease and related dementias for families, caregivers, and health professionals. ADEAR Center staff answer telephone, email, and written requests and make referrals to local and national resources.
What are the symptoms of Alzheimer's?
Common behavioral symptoms of Alzheimer’s include sleeplessness, wandering, agitation, anxiety, aggression, restlessness, and depression. Scientists are learning why these symptoms occur and are studying new treatments — drug and nondrug — to manage them.
What is the function of NMDAR?
1999, Monti and Contestabile 2000). This NMDAR-dependent neuroprotective functions mainly involves the activation of pro-survival transcription factors and the inhibition of apoptosis (Hardingham and Bading 2010, Parsons and Raymond 2014). The activation of synaptic NMDAR promotes survival gene expression by activating Ca2+-dependent transcription factors such as cyclic-AMP response element binding protein (CREB) and suppresses caspases and apoptotic pathway such as Puma (pro-apoptotic Bcl2 homology domain 3 (BH3)-only member gene) - activated signaling cascade and transcription factor FOXO (forkhead box protein O) - induced pro-death gene expression (Hardingham and Bading 2010, Parsons and Raymond 2014). The NMDAR-dependent synaptic plasticity and activation of cell survival signaling is illustrated in Figure 2.
What is the function of synaptic NMDAR?
Activation of synaptic NMDAR mediates survival pathway and induces LTP and LTD. In classical synaptic plasticity model, synaptic NMDAR activity activates CaMKII and phosphatases, which triggers LTP and LTD, respectively. Moreover, the activation of synaptic NMDARs suppresses the pro-apoptotic signaling molecules and pathways such as caspases, APAF1 and Puma occurred in cytoplasm; and it also promotes the expression of the pro-survival transcriptional factors such as CREB and suppresses the expression of pro-apoptotic transcription factors such as FOXO and p53, which, in turn, inhibits apoptosis and promotes cell survival.
What is the effect of glutamate on neuronal cell survival?
Insufficient synaptic NMDAR signaling compromises neuronal cell survival. Excessive stimulation of glutamatergic signaling, however, results in excitotoxicity, in which nerve cells are damaged or killed, or neurological trauma such as stroke (Rothman and Olney 1986). Besides acute effects, many studies indicate a role for glutamate excitotoxicity in delayed slowly-evolving neurodegeneration (Choi 1988, Lipton and Rosenberg 1994). Accumulating evidence demonstrates that the toxicity is principally mediated by excessive Ca2+entry, primarily through NMDARs (Choi 1987, Choi, Koh et al. 1988, Koh and Choi 1991, Tymianski, Charlton et al. 1993), since NMDARs have a much higher permeability for calcium ions compared to other iGluRs (Choi 1992). In this regard, the modest depolarization of the postsynaptic membrane and other factors that relieve the Mg2+blockade can activate NMDARs in a mild and chronic way, which causes the prolonged Ca2+influx into the postsynaptic neuron. The pathological level of Ca2+signaling leads to gradual loss of synaptic function and ultimate neuronal cell death, which correlates clinically with the progressive decline in cognition/memory and the development of pathological neural anatomy seen in AD patients, and this, in turn, rationalizes the clinical trial of memantine, an NMDAR antagonist, as a symptomatological and neuroprotective treatment for AD (Danysz, Parsons et al. 2000, Danysz and Parsons 2003, Wenk 2006). Thus, given the fact that NMDARs are also important for cell survival, the level of NMDAR signaling must be maintained at a proper level so that it is enough to promote neuronal survival but not harmful to cause neurodegeneration as occurred in AD. The major factors that affect NMDAR signaling in AD include glutamate availability and the modulation of NMDAR channel functions.
What is the role of glutamate receptors in neurotransmission?
These receptors also play fundamental roles in synaptic plasticity, the underlying molecular mechanism of learning and memory (Riedel, Platt et al. 2003). Owing to their pivotal roles in excitatory neurotransmission, the disruption of the normal signaling via iGluRs is implicated in a wide range of neuropathological disorders and diseases, such as epilepsy and brain damage, Parkinson’s, Alzheimer’s, Huntington’s, multiple sclerosis, thereby making iGluRs important drug targets for therapeutic purposes (Bleich, Romer et al. 2003).
What is glutamate in the brain?
Glutamate is the most abundant excitatory neurotransmitter in the mammalian Central Nervous System (CNS). It is extensively distributed in the CNS whereas it is almost exclusively located intracellularly. Glutamate can be synthesized through a number of metabolic pathways (Fonnum 1984). In wet tissue, glutamate is measured at concentrations of 5–15μmol/g (Perry, Yong et al. 1987, Erecinska and Silver 1990). Its concentration in the synaptic cleft at resting conditions is about 0.6μM (Bouvier, Szatkowski et al. 1992). During synaptic transmission glutamate concentration can go above 10μM at spatially localized extracellular regions (Clements, Lester et al. 1992). The residual glutamate is removed by glutamate uptake/transporter system (Danbolt 2001). The amount of available extracellular glutamate is subject to strict regulation to allow appropriate level of signaling.
What are the pathological changes associated with Alzheimer's disease?
The association of Alzheimer’s disease with Aβ, phosphorylated tau, mitochondria dysfunction and neuronal degeneration. Amyloid plaques and neurofibrillary tangles are commonly seen pathological changes in AD, which are formed from Aβ and phosphorylated tau, respectively. Aβ and amyloid plaques triggers oxidative stress and mitochondrial dysfunction, which damages synapses and promotes neuron degeneration. Neurofibrillary tangles aggravates these processes. The significant synaptic loss and neuronal death manifests the symptoms of Alzheimer’s disease.
Is glutamate a neurotransmitter?
The involvement of neurotransmitter glutamate and its receptors in the function of synaptic plasticity and the etiology of neurodegenerative diseases such as Alzheimer’s disease has been under investigation for many years. Recent studies reveal that glutamatergic neurotransmission through NMDARs lead to dichotomous results. Synaptic NMDAR signaling is required for the survival of neurons. However, extrasynaptic NMDARs signaling activated by the spillover of astrocyte- or presynaptic terminal-released glutamate plays a key role in antagonizing the synaptic pro-survival signaling pathway and tilt the balance toward excitotoxicity and ultimate neurodegeneration. This is supported by the beneficial clinical effects seen in moderate to severe AD cases by memantine, an FDA-approved NMDAR antagonist, which functions possibly through suppressing the extrasynaptic NMDAR signaling. Further studies on memantine and its derivatives will help elucidate the molecular mechanisms of how glutamate and NMDAR function in the etiology of AD.
What is the name of the drug used to treat Alzheimer's?
medication known as Namenda® (memantine), an N-methyl D-aspartate (NMDA) antagonist , is prescribed to treat moderate to severe Alzheimer’s disease. This drug’s main effect is to decrease symptoms, which could allow some people to maintain certain daily functions a little longer than they would without the medication. For example, Namenda® may help a person in the later stages of the disease maintain his or her ability to use the bathroom independently for several more months, a benefit for both the person with Alzheimer’s and caregivers.
What is the FDA's recommendation for a drug for Alzheimer's?
prescription drugs are currently approved by the U.S. Food and Drug Administration (FDA) to treat people who have been diagnosed with Alzheimer’s disease. Treating the symptoms of Alzheimer’s can provide people with comfort, dignity, and independence for a longer period oftime and can encourage and assist their caregivers as well.
What is the role of the N-methyl-D-aspartate receptor?
N -methyl- D -aspartate receptor plays a pivotal role in the synaptic transmission and synaptic plasticity thought to underlie learning and memory , which is not only central to the development and function of the nervous system, but also to neurotoxicity.
What is Alzheimer's disease?
Alzheimer’s disease (AD) is an age-related neurodegenerative disease. Clinically, this disorder is characterized by global cognitive dysfunction, especially memory loss, behavior and personality changes. AD progression has been associated with a gradual damage in function and structure in the hippocampus and neocortex, the vulnerable brain areas used for memory and cognition ( Mota et al., 2014 ). The neuropathologic hallmarks of AD include extracellular amyloid plaques, being composed of the amyloid beta (Aβ) peptide and intraneuronal neurofibrillary tangles (NFTs), consisting of abnormally hyperphosphorylated tau protein ( Ferreira et al., 2010 ). Nevertheless, damage and destruction of synapses seem to better correlate with memory loss than histopathologic markers. Synapse loss can be caused by the failure of live neurons to maintain functional axons and dendrites or by neuron death ( Bloom, 2014; Avila et al., 2017; Morris et al., 2018 ).
What are the functions of GluN2A and GluN2B?
In the adult CNS, particularly in higher brain structures, such as the hippocampus and cortex, GluN2A and GluN2B are the predominant subunits, indicating that they have central roles in synaptic function and plasticity ( Monyer et al., 1994; Takai et al., 2003 ). GluN2C- or GluN2D- containing receptors appear to give rise to ‘low-conductance’ openings with a lower sensitivity to extracellular Mg 2+, which may affect the Ca 2+ influx generated by synaptic activation of NMDAR ( Momiyama et al., 1996 ). Misra et al. (2000) results indicate that GluN1/GluN2D receptors do not contribute to the excitatory postsynaptic current (EPSC) and appear to be restricted to the extrasynaptic membrane. The possibility still exists that the GluN2D subunit is present at the synapse but is preferentially co-assembled with other GluN2 subunits, such as triheteromeric assemblies (GluN1/GluN2B/GluN2D) ( Dunah et al., 1998; Cull-Candy et al., 2001 ). GluN3A and GluN3B were cloned based on similarity to GluN1 and GluN2 subunits and were the last NMDA receptor subunits to be discovered ( Low and Wee, 2010 ). GluN3A expression is low embryonically, peaks during early postnatal life, and diminishes to lower levels in adulthood. This expression profile is observed in many regions of the brain ( Bendová et al., 2009; Roberts et al., 2009 ). Conversely, GluN3B levels are low around postnatal day 10 and gradually increase into adulthood within the neocortex, hippocampus, striatum, cerebellum, brainstem, and spinal cord ( Wee et al., 2008 ). GluN3A and GluN3B are expressed by multiple neuronal cell types, including interneurons, pyramidal cells, motor neurons, trigeminal neurons, retinal ganglion and amacrine cells ( Pachernegg et al., 2012 ). Although GluN3A is present in oligodendrocytes, it does not seem to be expressed in astrocytes ( Matsuda et al., 2003; Káradóttir et al., 2005; Salter and Fern, 2005 ). The GluN3 subunits bind glycine and D -serine ( Yao, 2006; Yao et al., 2008 ), but the functional properties and physiological roles of GluN3-containing NMDA receptors remain elusive. GluN3A acts in a dominant-negative manner to suppress receptor activity. GluN3A containing NMDARs display striking regional and temporal expression specificity, and, unlike most other NMDAR subtypes, they have a low conductance, are only modestly permeable to Ca 2+, and pass current at hyperpolarized potentials in the presence of magnesium. While glutamate activates triheteromeric NMDARs composed of GluN1/GluN2/GluN3A subunits, glycine is sufficient to activate diheteromeric GluN1/GluN3A-containing receptors ( Henson et al., 2010 ). About GluN3A is expressed in the right place at the right time to regulate spine and synapse development, there are two hypotheses, if the influence of GluN3A is to serve as a ‘synaptic brake’ to limit synapse/spine formation or if it serves as a ‘synaptic marker’ to promote the elimination of spines ( Henson et al., 2010 ). Roberts et al. (2009) found that prolonging GluN3A expression prevents glutamatergic synapse maturation by limiting synapse potentiation and growth, and decreasing spine density, accompanied by major impairments in learning and memory processes. Whereas knocking out endogenous GluN3A conversely accelerates synaptic maturation events.
How many NMDAR subunits are there?
Seven different NMDAR subunits have been identified: the GluN1 subunit, four distinct GluN2 subunits (GluN2A, GluN2B, GluN2C, and GluN2D), and a pair of GluN3 subunits (GluN3A and GluN3B) ( Paoletti, 2011 ). Functional NMDARs are heterotetramers composed of two glycine or D -serine-binding GluN1 subunits and two glutamate-binding GluN2 (GluN2A-D) subunits or, in some cases, glycine-binding GluN3 (GluN3A/B) subunits ( Köhr, 2006 ). All NMDAR subunits share a common membrane topology that contain four discrete domains ( Lee et al., 2014; Karakas and Furukawa, 2014 ): The extracellular amino-terminal domain (ATD) is involved in subunit assembly and allosteric regulation, and contributes to control of ion channel open probability and deactivation speeds. The extracellular ligand-binding domain (LBD) is formed by two discontinuous segments (S1 and S2), and binds agonists and antagonists to control ion channel opening. The transmembrane domain (TMD) contains three transmembrane segments (M1, M3, and M4) and a re-entrant pore loop (M2) which is part of the channel pore, containing a critical asparagine residue that determines calcium permeability of the channel and mediates the magnesium blockade ( Paoletti et al., 2013; Karakas and Furukawa, 2014 ). The intracellular carboxyl-terminal domain (CTD) interacts with multiple cytosolic proteins ( Sanz-Clemente et al., 2012 ). GluN2 LBDs contain the glutamate binding site, while GluN1 (or GluN3) LBDs contain the Glycine/ D -serine coagonist binding site ( Hackos and Hanson, 2017 ). In general, activation of NMDARs containing GluN1/GluN2 requires binding of both glycine and glutamate at the extracellular LBDs in addition to release of magnesium block by membrane depolarization at the TMD ( Regan et al., 2015 ). Positively charged residues on the surface of the GluN2A LBD assist glutamate binding via a ‘guided-diffusion’ mechanism. On the other hand, Glycine binds to the GluN1 LBD via an ‘unguided-diffusion’ mechanism, whereby glycine finds its binding site primarily by random thermal fluctuations ( Yu and Lau, 2018 ). A recent study show, through cryoelectron microscopy, that the presence of the exon 5 motif, at the ATD of the GluN1 subunit, alters the local architecture of heterotetrameric GluN1/GluN2 NMDA receptors and creates contacts with the LBDs of the GluN1 and GluN2 subunits. The unique interactions established by the exon 5 motif are essential to the stability of the ATD/LBD and LBD/LBD interfaces that are critically involved in controlling proton sensitivity and deactivation ( Regan et al., 2018 ). The most widely expressed NMDARs contain the obligatory subunit GluN1 plus either GluN2B or GluN2A or a mixture of the two; therefore, in next sections we will focus on these two subunits.
Where is GluN2A expressed?
During the first two postnatal weeks, GluN2A expression rises steadily to become widely and abundantly expressed in virtually every CNS area in the adult. Meantime, GluN2D expression drops markedly, and in the adult, it is expressed at low levels mostly in the diencephalon and mesencephalon.
Which receptors are sensitivity to glycine?
The sensitivity to glycine, conferred by the GluN1 sub-units, is close to that of GluN1/GluN2B receptors, whereas the sensitivity to glutamate, conferred by the GluN2 sub-units, is closer to that of GluN1/GluN2A receptors ( Hansen et al., 2014; Stroebel et al., 2014 ).
Does glutamate activate extrasynaptic NMDARs?
Certain interactions of NMDAR subunits with distinct signaling molecules may occur at synaptic but not at extrasynaptic sites ( Köhr, 2006 ). Extrasynaptic NMDARs are exposed to ambient glutamate, whether this glutamate concentration is high enough to tonically activate extrasynaptic NMDARs remains controversial. Although microdialysis studies report that ambient glutamate concentrations in vivo are high enough to activate extrasynaptic NMDARs ( Nyitrai et al., 2006 ), a study suggests that glutamate transporters regulate ambient glutamate concentrations at a level that is too low to cause significant receptor activation ( Herman and Jahr, 2007 ). While, some reports that glutamate that is released into the extracellular space mainly from glial processes ( Fellin et al., 2004) may result in the persistent activation of extrasynaptic GluN2B receptors, which are of high affinity and are sensitive to low concentrations of glutamate ( Vizi, 2000 ).
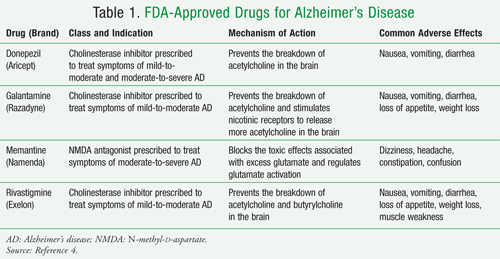
What is NMDA antagonist?
NMDA antagonist and cholinesterase inhibitor prescribed to treat symptoms of moderate to severe Alzheimer’s
What is the best treatment for Alzheimer's?
disease-modifying drugs or therapies. Aducanumab is the only disease- modifying medication currently approved to treat Alzheimer’s. This medication is a human antibody, or immunotherapy, that targets the protein beta-amyloid and helps to reduce amyloid plaques, which are brain lesions associated with Alzheimer’s. Aducanumab was granted accelerated approval by the FDA based on its ability to reduce amyloid in the brain but not on the basis of any demonstrated effect on slowing progression of cognitive decline or dementia. Researchers are continuing to study whether this medication works to affect a person’s rate of cognitive decline over time. Before prescribing aducanumab,
What is the effect of N-methyl D-aspartate?
an N-methyl D-aspartate (NMDA) antagonist, is prescribed to treat moderate to severe Alzheimer’s disease. This drug’s main effect is to decrease symptoms, which could enable some people to maintain certain daily functions a little longer than they would without the medication. For example, memantine may help a person in the later stages of the disease maintain his or her ability to use the bathroom independently for several more months.
What is a cholesterase inhibitor?
Cholinesterase inhibitor prescribed to treat symptoms of mild, moderate, and severe Alzheimer's
What are the symptoms of Alzheimer's?
Alzheimer’s include sleeplessness, wandering, agitation, anxiety, aggression, restlessness, and depression. Scientists are learning why these symptoms occur and are studying new treatments — drug and nondrug — to manage them. Research has shown that treating behavioral symptoms can make people with Alzheimer’s more comfortable and makes things easier for caregivers. Experts agree that medicines to treat these behavior problems should be used
What is disease modifying immunotherapy?
Disease-modifying immunotherapy prescribed to treat mild cognitive impairment or
Which disease is associated with amyloid plaques?
Neurons in the brain surrounded by amyloid plaques, which are associated with Alzheimer's disease.