Is there a cure for Pompe disease?
Aug 09, 2021 · The The U.S. Food and Drug Administration has approved alglucosidase alfa (Myozyme©) to treat infantile-onset Pompe disease. Lumizyme© have has been approved to treat individuals of all ages with Pompe disease. The drug avalglucosidare alfa-ngpt (Nexviazyme©) has been approved for i... View Full Treatment Information Prognosis
Who treats Pompe disease?
Enzyme Replacement Therapy. Currently, the only available, specific, and effective treatment for Pompe disease is enzyme replacement therapy (ERT), 3 which was first approved in 2006 for use in patients with infantile-onset Pompe disease (IOPD). In ERT, synthetic recombinant human GAA (rhGAA) is administered intravenously every 2 weeks to replace low levels of GAA in affected …
How does Myozyme treat Pompe disease?
How is Pompe disease treated? Enzyme replacement therapy (ERT) is an approved treatment for all Pompe patients. A drug called alglucosidase alfa is given intravenously (through the patient’s vein). It is a genetically engineered enzyme that acts like the naturally occurring acid alfa glucosidase enzyme.
How is Pompe disease treated?
Aug 10, 2021 · In the U.S., the approved ERTs for Pompe disease are Lumizyme (alglucosidase alfa) and the next-generation therapy Nexviazyme (avalglucosidase alfa). Both Pompe disease treatments are manufactured by Sanofi Genzyme. Lumizyme, marketed as Myozyme, also has been approved in the European Union. Supportive therapies
How long can you live with Pompe disease?
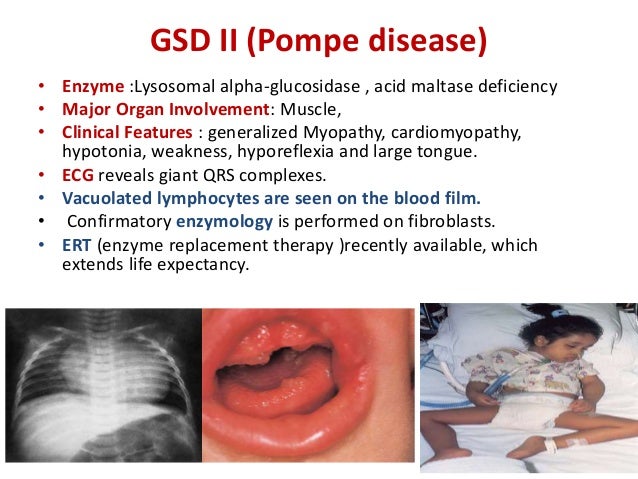
They have characteristic heart (cardiac) problems (dysfunction due to heart enlargement) in addition to generalized skeletal muscle weakness and a life expectancy of less than 2 years, if untreated (classic infantile Pompe disease).
What happens to people with Pompe disease?
Pompe disease happens when your body can't make a protein that breaks down a complex sugar, called glycogen, for energy. Too much sugar builds up and damages your muscles and organs. Pompe disease causes muscle weakness and trouble breathing.Jun 3, 2020
Is Pompe disease painful?
The median pain severity score in Pompe patients reporting pain was 3.1 (on a scale from 0 to 10), indicating mild pain; against 2.6 amongst controls (p=0.06). The median score of pain interference with daily activities in patients who reported pain was 3.3, against 1.3 in controls (p=0.001).
Can you live a normal life with Pompe disease?
They can survive up to age 30 if the disease appears in childhood and up to age 50 if it develops in adulthood. Generally, the later the age of onset, the slower the disease progression and the longer the life expectancy.Oct 28, 2019
Is Pompe always fatal?
What Is the Life Expectancy for Pompe Disease? The infantile form of Pompe disease (type II glycogen storage disease) is usually fatal, and most patients die within 1 year of birth. Enlarged heart with progressive obstruction to left ventricular outflow is a major cause of death.Jun 26, 2020
Is Pompe disease fatal?
Pompe disease is a rare (estimated at 1 in every 40,000 births), inherited and often fatal disorder that disables the heart and skeletal muscles. It is caused by mutations in a gene that makes an enzyme called acid alpha-glucosidase (GAA).Aug 9, 2021
Who gets Pompe?
Since this is a genetic condition, the people who get this disease inherit it from a parent. It is common, however, that neither parent shows any symptoms. The disease is rare. In the United States, only 1 person in 40,000 is affected by Pompe disease.Apr 2, 2019
Is McArdle disease fatal?
The disease can lead to dark urine. Severe, uncontrolled McArdle disease can cause life-threatening kidney problems.
Do both parents have to be carriers for Pompe disease?
Even when both parents have the mutated gene, all their children won't necessarily get Pompe disease, or be carriers for it. When both parents are Pompe disease carriers, babies inherit two working GAA genes 25% of the time. They inherit two nonworking GAA genes -- which leads to Pompe disease -- 25% of the time.Jan 19, 2022
What is the mortality rate of Pompe disease?
Survival from diagnosis The estimated 5-year survival after diagnosis was 95%. At 10, 20 and 30 years this was 83, 65 and 40%, respectively (Figure 1). Survival estimates of 268 untreated adults with Pompe disease from diagnosis until end of study, start of ERT or death. Twenty-three patients died during follow-up.Jun 1, 2011
How common is it to be a carrier of Pompe disease?
If both parents have Pompe disease, then every child will inherit the disease. If one parent has the disease and the other is a carrier, each child has a 50% chance of inheriting the disease and a 50% chance of being a carrier.
Treatment
Pompe disease is a multi-system disorder caused by mutations in the GAA gene that codes for the enzyme, acid alpha-glucosidase (GAA), 1 which breaks down glycogen into glucose to be used as an energy source for the cells. Defects in the enzyme prevent glycogen breakdown, causing a toxic accumulation of glycogen inside the cells.
Enzyme Replacement Therapy
Currently the only available, specific, and effective treatment for Pompe disease is enzyme replacement therapy (ERT), 3 which was first approved in 2006 for use in patients with infantile-onset Pompe disease (IOPD).
Limitations of ERT
ERT has improved cardiac function and extended survival times in patients with IOPD. However, patients are not fully cured and residual symptoms remain. Respiratory and swallowing functions are only stabilized. About 30% of patients who received ERT still require assisted ventilation. 5
Future ERT Studies
To overcome the limitations of rhGAA therapy, modified rhGAA (avalglucosidase alfa, Neo-GAA) with a higher affinity for M6P receptors is being investigated in a Phase 3 clinical study (NCT02782741). 6
What is Pompe disease?
Pompe disease is a genetic disorder in which complex sugar called glycogen builds up in the body’s cells. The disease results from the deficiency of an enzyme called acid alfa glucosidase (GAA), which breaks downs complex sugars in the body. This buildup occurs in organs and tissues, especially in muscles, causing them to break down.
How many types of Pompe disease are there?
What are the types of Pompe disease? There are three types of Pompe disease: Classic infantile-onset appears within a few months of birth. Non-classic infantile-onset appears at about 1 year of age. Late-onset appears later in a child’s life, or even into the teen years or adulthood.
What are the symptoms of a baby with a swollen liver?
In infants, symptoms include the following: Classic type: Weak muscles. Poor muscle tone. Enlarged liver. Failure to gain weight and grow at the expected rate (failure to thrive) Trouble breathing. Feeding problems.
Can a baby die from Pompe disease?
Without treatment, infants with Pompe disease will die. Many of the people with Pompe disease have respiratory (breathing) problems, heart problems, and almost all are plagued with muscle weakness. Most people will have to use oxygen and wheelchairs at some point.
What is a blood sample?
A blood sample is taken and enzymes in the blood are studied and counted. Also, there are tests such as sleep studies, breathing tests to measure lung capacity, and electromyography (a test that measures how well the muscles work). A blood sample is taken and enzymes in the blood are studied and counted.
What tests are done to determine lung capacity?
Breathing tests to measure lung capacity ( pulmonary function tests) Electromyography (a test that measures how well the muscles work) and MRI s. Heart studies, including X-rays, electrocardiogram and echocardiogram. Sleep studies. A prenatal diagnosis may be done for pregnant women at risk.
What is ERT in Pompe?
Enzyme replacement therapy (ERT) is an approved treatment for all Pompe patients. A drug called alglucosidase alfa is given intravenously (through the patient’s vein). It is a genetically engineered enzyme that acts like the naturally occurring acid alfa glucosidase enzyme.
What is the best treatment for Pompe disease?
Supportive therapies. Most patients require supportive therapy to address the symptoms of Pompe disease, which include respiratory and cardiac problems, physical disability, and difficulty swallowing. Some patients may require a mechanical ventilator during respiratory infections or at night. Physical and occupational therapy can improve patients’ ...
What is the cause of Pompe disease?
Pompe Disease Treatments. The symptoms of Pompe disease are caused by missing or insufficient amounts of an enzyme called acid alpha-glucosidase (GAA). This enzyme is needed to break down glycogen, a complex sugar molecule, into glucose, the simple sugar that our bodies use for energy. If glycogen is not turned into glucose, ...
What is ERT in medicine?
Enzyme replacement therapy. Enzyme replacement therapy (ERT) is used to increase the levels of GAA in the body and reduce the accumulation of glycogen inside cells. The enzyme is made in the laboratory using cultured cells or cells grown and maintained in vitro, which have been genetically altered to produce the human form of GAA.
Does ERT help with Pompe disease?
ERT is administered into the bloodstream and has been shown to reduce the abnormal thickening of the heart walls that is characteristic of Pompe disease. It also helps maintain muscle function and improve patients’ quality of life.
Approved Treatments
Enzyme replacement therapy is the current standard treatment for patients with Pompe disease. This approach is used to increase the levels of GAA in the body and reduce the accumulation of glycogen inside cells. Lumizyme (alglucosidase alfa) is the only approved ERT for Pompe in the U.S. It is available in the EU under the brand name Myozyme.
Experimental Treatments
There are a number of experimental treatments for Pompe disease in various stages of development. These treatments are not yet approved, but they offer the potential for more options and better outcomes for Pompe patients in the future.
Non-drug Treatments
Most patients require supportive therapy to address the symptoms of Pompe disease, which include respiratory and cardiac problems, physical disability, and difficulty swallowing.
Treatment Options
Pompe disease being a genetic disorder, screening for the common genetic mutations helps confirm the diagnosis. Measuring the GAA enzyme level in blood also plays an important role in correct diagnosis of the disease. An expert geneticist can help detect the carriers with the help of genetic mutation analysis.
Symptoms
Statistics show that in the U.S., about one in 40,000 people is affected by the Pompe disease. It is considered as a fatal disease as it leads to dysfunction of the heart and muscles. Mutations in a gene results in Pompe disease. Build up of glycogen everywhere in the body leads to several difficulties.
What are the symptoms of Pompe disease?
Symptoms of late-onset Pompe disease might include: 1 Weakness in the truck, legs, or arms 2 Lung infections 3 Shortness of breath and breathing troubles during sleep 4 Spine curvature 5 Enlarged liver 6 Enlarged tongue, which can affect chewing and swallowing 7 Stiff joints
How many people have Pompe disease?
Pompe disease affects one in 40,000 people in the United States. 1 It is an inherited condition caused by gene mutations in the gene that makes an enzyme called acid alpha-glucosidase (GAA). This condition belongs to a group of disorders called lysosomal storage disorders.
What is ERT used for?
ERT is also used to treat Fabry disease and Gaucher disease. Like Pompe disease, these two conditions are lysosomal storage disorders. With ERT, alpha-glucosidase is infused directly into the bloodstream, where the body responds to it and breaks down glycogen to prevent a toxic buildup in the cells.
How does ERT work?
Additional Treatments. Enzyme replacement therapy (ERT) is an effective treatment for Pompe disease. It involves injecting alpha-glucosidase directly into the bloodstream. ERT helps the body to break down glycogen and prevents its toxic buildup. It will also alleviate symptoms and slow down the progression ...
Can ERT be stopped?
Once started, ERT is rarely stopped even if a person experiences negative side effects. ERT is linked to improved strength and energy levels, as well as higher survival rates. 6 Unfortunately, ERT cannot resolve neurological symptoms and effects of Pompe disease.
What are the symptoms of hypoventilation?
Mild symptoms of hypoventilation include tiredness, daytime sleepiness, shortness of breath, slow or shallow breathing, and depression. As the condition worsens and carbon dioxide levels go up, a person might experience bluish color of lips, fingers, or toes, headaches, confusion, and/or seizures.
What are the symptoms of anaphylaxis?
Symptoms of anaphylaxis might include feeling lightheaded or faint, fast or shallow breathing or other breathing difficulties, wheezing, rapid heartbeat, confusion, anxiety, clammy skin, and loss of consciousness. 9
What is the cause of Pompe disease?
What Is Pompe Disease? Pompe disease results when mutations occur in the gene that triggers the production of an enzyme called acid alpha-glucosidase (GAA). That enzyme is responsible for helping the body break down glycogen (sugar).
How often is myozyme given?
Given via four-hour intravenous infusions every two weeks for the rest of a patient's life, Myozyme successfully improves heart and muscular function. With its use, children are able to meet their motor milestones, they are running, going to school and playing games.".
When did myozyme start?
Myozyme's journey started in 1995 when Chen engineered a line of cells that could produce the GAA enzyme. Within one year, animal studies proved fruitful when birds missing the enzyme went from being unable to fly to flipping from their backs to their feet after several days of infusions. One of the birds even flew.
What is the movie Extraordinary Measures about?
When the film Extraordinary Measures debuts on January 22, it will tell the story of one man's quest to obtain treatment for his children who suffer from a rare metabolic disorder called Pompe disease. The real story began 20 years ago at Duke University Medical Center when pediatric geneticist ...